Research Directions
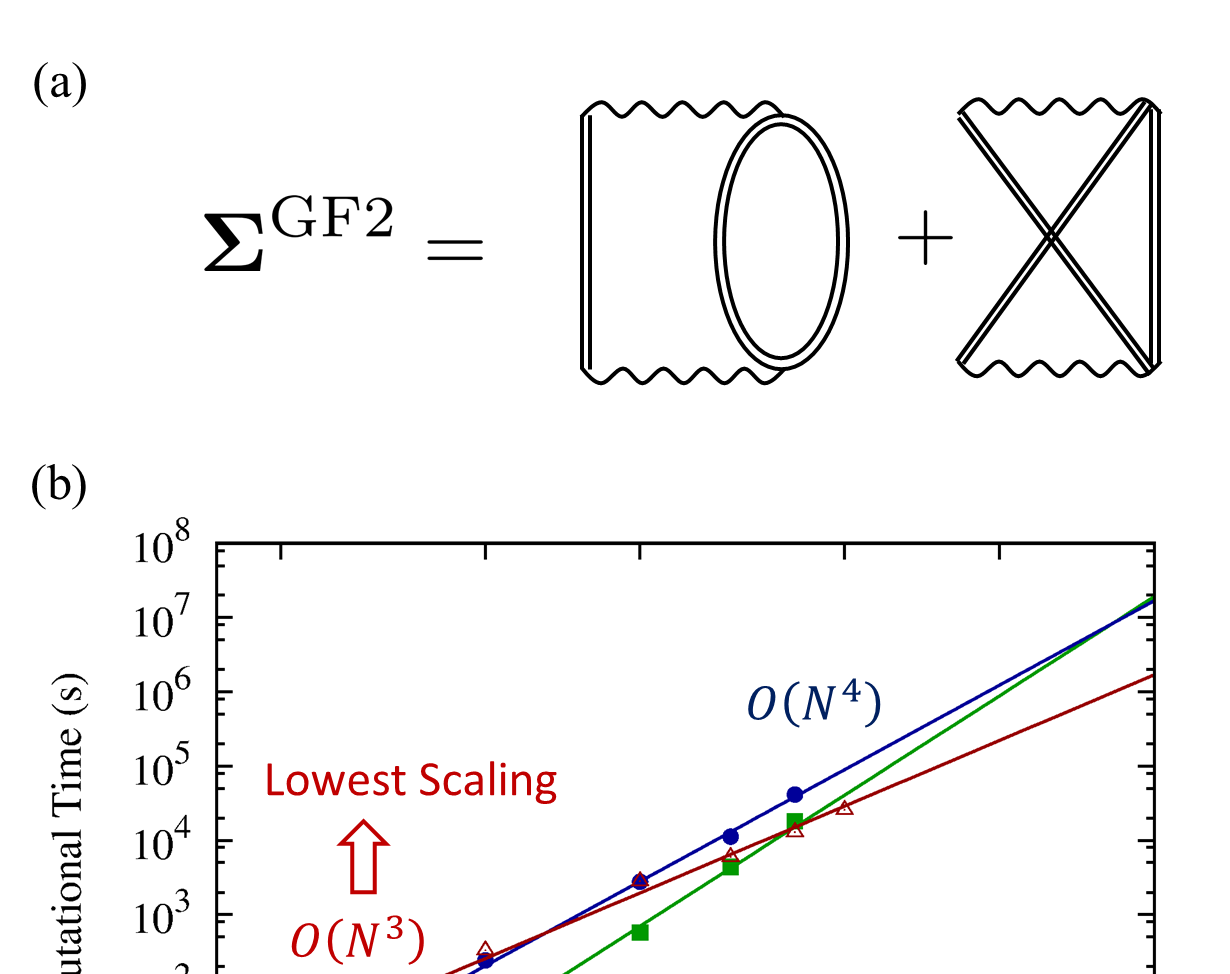
Methods Development
- Electronic structure
- Electronic dynamics
- Many-body Green's functions
- Stochastic methods
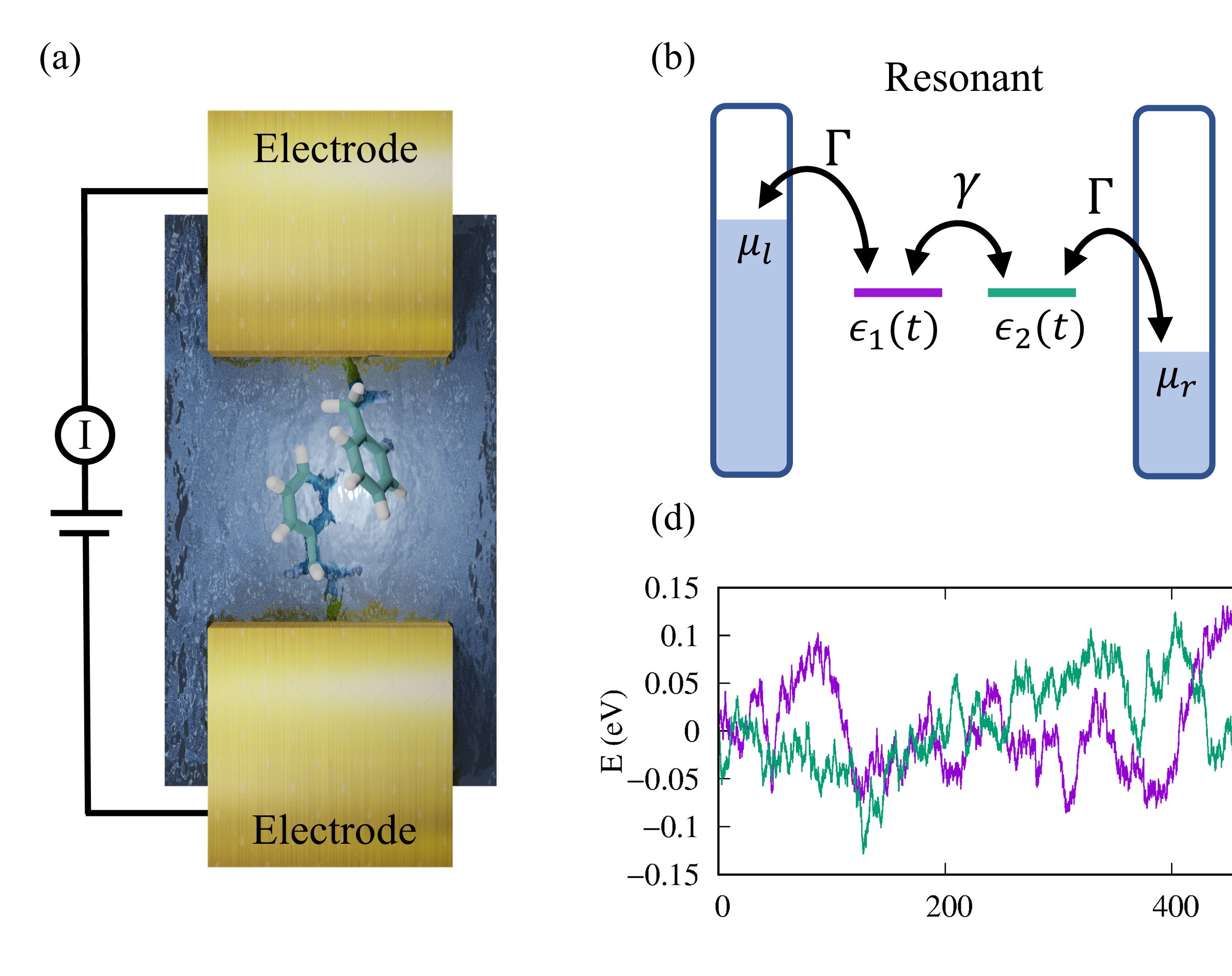
Nano-Electronics
- Charge and spin transport
- External stimuli control
- Applications in nanoelectronics and spectroscopy
Nanostructures & Materials
- Electronic structure (band structure)
- Quasiparticles
- Light-matter interactions






